Gene Expression
Nebulas
GENtoolkit provides powerful pipelines which can handle both bulk and single-cell (10X Genomics, Smart-seq2, Drop-seq and inDrop) RNA-seq data. All gene/transcript expression profiles deposited in Gene Expression Nebulas are processed based on GENtoolkit. GENtoolkit is composed of two main parts which correspond to upstream and downstream analysis pipelines respectively. Specifically, upstream analysis module includes 4 steps, 'index building', 'quality control', 'read alignment', 'gene expression quantification', while downstream analysis module includes 2 main steps, 'analysis of gene expression profiles' and 'visualization of analysis results'. Raw data in the format of 'sra' or 'fastq' (single-end or paired-end) are both supported for further gene/transcript expression profiling. According to the needs of users, it is accessible to perform gene expression analysis in all or part samples from a dataset.
tar -zvxf GENtoolkit.tar.gz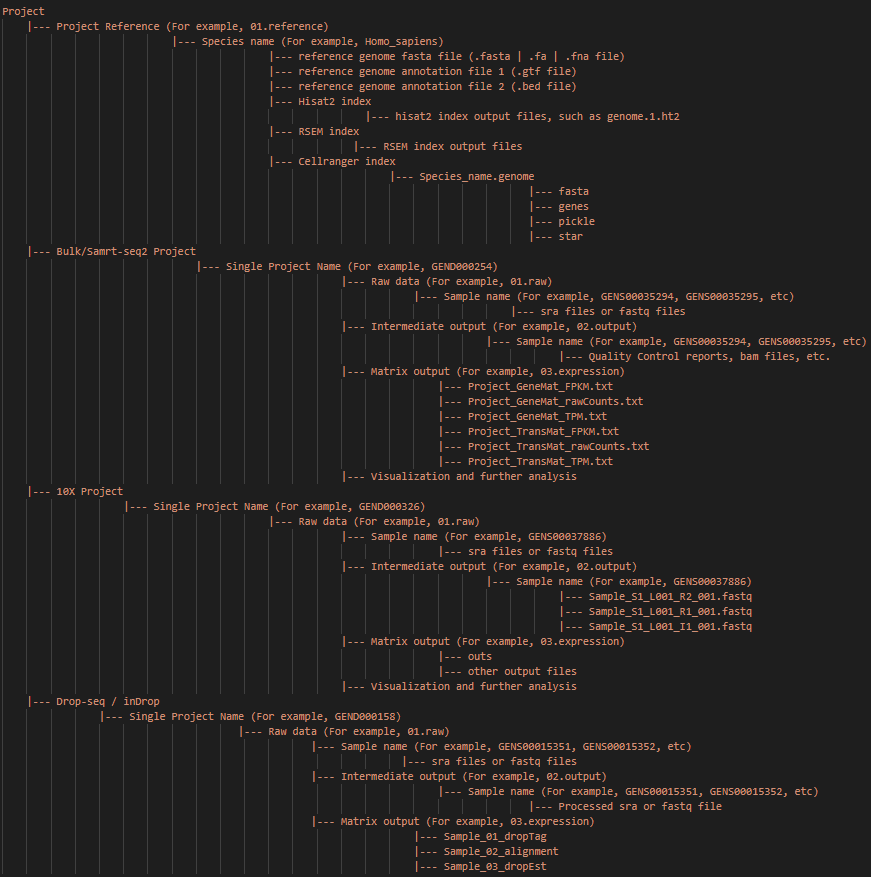
Note: The main input and output files and paths of the whole project are shown above. And this directory structure is recommended.
python GENtoolkit.py [options] ...python GENtoolkit.py [options] -blt Bulk -ipp
../Oryza_sativa -rgf reference.fasta -rgg reference.gtf -sp
../STAR/bin/Linux_x86_64 -rd ../01.raw -st pair -hi
../Oryza_sativa_hisat2/genome -ri
../Oryza_sativa_RSEM/Oryza_sativa -bf reference.bed
Note: Test data comes from GEN (GEND000254 and GEND000257)
python GENtoolkit.py [options] -blt Smart-seq2 -ipp ../Oryza_sativa -rgf reference.fasta -rgg reference.gtf -sp ../STAR/bin/Linux_x86_64 -rd ../01.raw -st pair -hi ../Oryza_sativa_hisat2/genome -ri ../Oryza_sativa_RSEM/Oryza_sativa -bf reference.bed
Note: Test data comes from GEN (GEND000305)
python GENtoolkit.py [options] -blt 10X -ipp ../Saccharomyces_cerevisiae -rgf reference.fasta -rgg reference.gtf -sp ../STAR/bin/Linux_x86_64 -rd ../01.raw -ci ../Saccharomyces_cerevisiae/Saccharomyces_cerevisiae.genome
Note: Test data comes from GEN (GEND000326). If some samples run failed because of the limitation of cores, or mem, or something else, "-da Designated_samples -sl sample1-sample2-sample3" can help re-run.
python GENtoolkit.py -blt Drop-seq -ipp ../Homo_sapiens -rgf reference.fasta -rgg reference.gtf -si ../STAR/bin/Linux_x86_64 -dc reference.xml -dm ../mit_genes_human.ensembl.rds -rd ../01.raw
Note: Test data comes from GEN (GEND000158). If "-da Designated_samples" selected, the parameter "-sl []" is necessary!
python GENtoolkit.py [options] --stream down --BuildLibraryType Bulk --workpath ./workpath/ --exprData exprData.txt --metaData metaData.txt
python GENtoolkit.py [options] --stream down --BuildLibraryType Smart-seq2 --workpath ./workpath/ --exprData exprData.txt --metaData metaData.txt -- refpath ./ref/
python GENtoolkit.py [options] --stream down --BuildLibraryType 10X --workpath ./workpath/ --exprData ./data/ (including barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz) -- refpath ./ref/
| Long parameter | Short parameter | Description |
|---|---|---|
| --stream | -stream | Pipeline type, [ up | down | all ] |
| --IndexBuild | -ib | Once a reference genome is built, it can be used many times in a species gene expression analysis, which depends on whether building index or not. [ index_build | index_exist ] Default: index_build |
| --BuildLibraryType | -blt | Library building type, [ Bulk | 10X | Smart-seq2 | inDrop | Drop-seq ] |
| --IndexProjectPath | -ipp | The absolute path of the index-building project (Species name). |
| --Hisat2ThreadNum | -htn | The thread number for hisat2 index-building. Default: 30 |
| --RSEMThreadNum | -rtn | The thread number for RSEM index-building. Default: 30 |
| --ReferenceGenomeFasta | -rgf | The fasta file of reference genome. |
| --ReferenceGenomeGtf | -rgg | The gtf file of reference genome. |
| --StarPath | -sp | The absolute path of the software STAR. |
| --DesignatedAll | -da | The sample list you want to run at once. [ Designated_samples | All_samples ] Default: All_samples |
| --SampleList | -sl | The list samples you designated. [ Sample1-Sample2-Sample3 ] Note: The short dashes are needed. |
| --SequencingType | -st | [ pair | single ] |
| --ReadType | -rt | [ sra | fastq ] Default: fastq |
| --RawData | -rd | The absolute path of raw data |
| --Hisat2Index | -hi | The Hisat2 reference genome index, for example, ../Homo_Sapiens_hisat2/genome |
| --RSEMIndex | -ri | The RSEM reference genome index, for example, ../Homo_Sapiens_RSEM/Homo_Spaiens |
| --BedFile | -bf | The browse extensive data file (.bed file). |
| --FasterqDumpThread | -fdt | The thread number for fasterq-dump. Default: 12 |
| --Fastp_q | -fq | The parameter "-q" in fastp. Default: 20 |
| --Fastp_u | -fu | The parameter "-u" in fastp. Default: 20 |
| --Fastp_l | -fl | The parameter "-l" in fastp. Default: 50 |
| --Fastp_W | -fW | The parameter "-W" in fastp. Default: 4 |
| --Fastp_M | -fM | The parameter "-M" in fastp. Default: 12 |
| --Fastp_w | -fw | The parameter "-w" in fastp. Default: 12 |
| --Hisat2_p | -hp | The thread number for hisat2. Default: 12 |
| --SamtoolsThread | -std | The thread number for samtools. Default: 12 |
| --RSEMThread | -rtd | The thread number for RSEM. Default: 12 |
| --CellrangerIndex | -ci | The absolute path of cellranger index, for example, ../Homo_Sapiens/Homo_Sapiens.genome |
| --CellrangerLocalCores | -clc | Caution! The default localcores may not be appropriate all the time, you can adjust the localcores according to https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/count. Default: 12 |
| --CellrangerLocalMem | -clm | Caution! The default localmem may not be appropriate all the time, you can adjust the localmem according to https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/count. Default: 64 |
| --DropTag_p | -dp | The thread number of dropTag process. Default: 12 |
| --StarRunThreadN | -srtn | The thread number of STAR alignment. Default: 12 |
| --DropEst_c | -dc | The configure file (.xml file) of DropEst process. |
| --DropReport_m | -dm | The reference organelle gene's rds file. |
| Long parameter | Short parameter | Description |
|---|---|---|
| --workpath | -wp | The absolute path of work path. |
| --exprData | -exp | Expression matrix file, if stream is "all", it would use upstream results. |
| --metaData | -md | Meta information file. |
| --refpath | -rf | Reference file for single cell data. |
| --geneList | -gl | Gene list file. |
| --thread | -td | Number of thread for downstream. Default: 10 |
| --picType | -pic | Picture file format, [ svg | pdf | png ]. Default: svg |
| --rowSums_count_cutoff | -rc | Low count filter. The gene under rowSums count cutoff in samples would be deleted. Default: 2 |
| --pValue_cutoff | -pc | The p value cutoff in diferent analysis. Default: 0.05 |
| --padj_cutoff | -qc | The q value cutoff in diferent analysis. Default: 0.1 |
| --log2FoldChange_Up | -F | Log2(FoldChange) in up regulation cutoff. Default: 1 |
| --log2FoldChange_Down | -f | Log2(FoldChange) in down regulation cutoff. Default: -1 |
| --topnum | -tn | Top number gene cutoff. Using how many top gene as result. Default: 20 |
| --module | -module | Select module to export from colorlevels in WGCNA. Default: turquoise |
| --outputType | -ot | Select export format, [ VisANT | cytoscape ]. Default: cytoscape |
| --nTop | -ntop | Export network cutoff: the number of the top gene to be filtered and exported in selected module. Default: 10000 |
| --NetworkThreshold | -nt | Export Network cutoff: weight > NetworkThreshold. Default: 0.02 |
| --GeneSign | -gs | Hub gene cutoff: significance of single gene-trait correlation > GeneSign. Default:0.1 |
| --absdatKME | -KME | Hub gene cutoff: eigengene connectivity (kME) value > absdatKME Default:0.1 |
| --qWeight | -qw | Weight cutoff. Default:0.9 |
| --GOAnalysis | -GO | GO enrichment analysis, [ yes | no ]. Default: yes |
| --KEGGAnalysis | -KEGG | KEGG enrichment analysis, [ yes | no ]. Default: yes |
| --DOAnalysis | -DO | DO enrichment analysis, [ yes | no ]. Default: yes |
| --ontology | -ontology | GO Ontology of GO enrichment analysis, [ ALL | BP | MF | CC ]. Default: ALL |
| --pValue_cutoff_enrich | -pen | Enrichment analysis cutoff to p value. Default:0.05 |
| --padj_cutoff_enrich | -qen | Enrichment analysis cutoff to q value. Default:0.1 |
| --Biological_Condition | -bc | Choose one case or control, consistent with meta table. Default:Case1 |
| --TreeMethod | -tm | TreeMethod, default, [RF (Random Forests) | ET (Extra-Trees)]. Default: RF |
| --nTrees | -ntrees | Number of trees in an ensemble for each target gene. Default:50 |
| --min_cells | - | The minimum number of cells. Default:5 |
| --max_cells | - | The maximum number of cells. Default:10000 |
| --min_genes | - | The minimum number of genes. Default:200 |
| --max_genes | - | The maximum number of genes. Default:4000 |
| --max_mito | - | The maximum number of mito. Default:0.05 |
| --PCnumber | - | The top PC number for find Neighbor. Default:15 |
| --min_Resolution | - | The minimum Resolution for pre-find cluster. Default:0.6 |
| --max_Resolution | - | The maximum Resolution for pre-find cluster. Default:1.2 |
| --intervals_Resolution | - | The Resolution for pre-find cluster. Default:0.2 |
| --Resolution | - | The Resolution for find cluster. Default:1 |
| --method | - | [ tsne | umap ]. Default:tsne |
| --whichCluster | - | Choose one cluster for find marker and show on TSNE plot, the first cluster label as 0 . Default:1 |
| --ref | - | Choose a reference, default [ 1-6 ], 1 HumanPrimaryCellAtlasData, 2 BlueprintEncodeData, 3 MonacoImmuneData, 4 NovershternHematopoieticData, 5 DatabaseImmuneCellExpressionData, 6 HumanPrimaryCellAtlasData and BlueprintEncodeData. Default:1 |
| --annolevel | - | [ main | fine ]. Default:main |