

ParaAT (Parallel Alignment and back-Translation) is a parallel tool for constructing protein-coding DNA alignments for a large number of homologous groups.
Alignments of protein-coding DNA sequences are more informative than their corresponding protein alignments and accordingly are more useful for a large variety of bioinformatic analyses. Due to the evolution of coding sequences as triplets of nucleotides, the larger alphabet of amino acid (than that of nucleotide), and the relative conservation of amino acid sequences (in contrast to their DNA counterparts), back-translated nucleotide alignments guided by amino acid alignments are more reliable and accurate than direct nucleotide alignments.
With the exponentially growing amount of sequence data available and the ever-ongoing efforts to dig out the “treasure” from big data which are primarily reliant on multiple protein-coding DNA alignments, ParaAT is capable of processing a large number of homologs and constructing multiple protein-coding DNA alignments.
ParaAT provides good scalability and achieves high parallel efficiency for computationally demanding tasks, and thus is of great usefulness for large-scale data analysis in the high-throughput era.
No restriction on the number of homologous groups; it is dependent on the memory size of computer used. In addition, different rows (groups) can have different numbers of homologous genes.
The number of processors is provided as a file input for ParaAT instead of a command input.
ParaAT reads the number of processors (N) from the corresponding input file, as soon as N tasks are complete. That is to say, the number of processors is read from the file frequently, which can make the number of processors changeable during running time.
ClustalW, Mafft, Muscle and T-Coffee.
We tested on two datasets: 15,430 pairs of human-mouse homologs and 9,712 trios of human-chimp-mouse homologs. The test was performed on a 64 bit x86 Intel® Xeon® machine with 24 cores (X5650) at 2.67 GHz each running CentOS release 6.1. We recorded the running time of ParaAT to construct multiple protein-coding DNA alignments for both datasets using 1–16 CPUs.
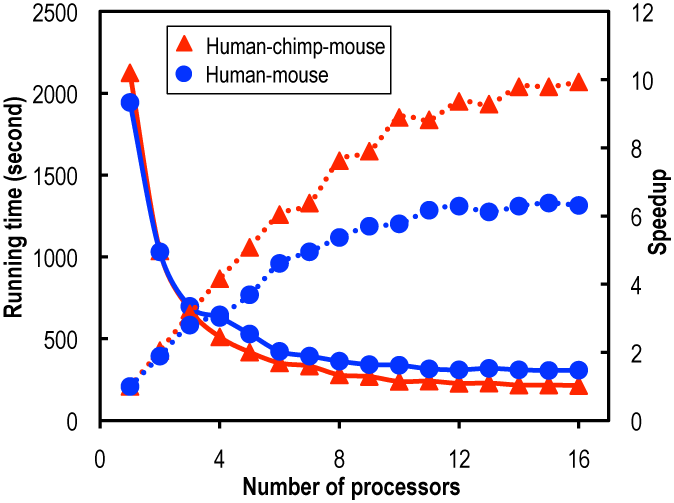
As the number of processors increases, the speedup (defined as a ratio of the running time with one processor to that with p processors) is dramatically on the rise and almost linear for up to about eight processors, indicating good scalability. Across all numbers of processors examined, the speedup in human-chimp-mouse trios is always better than that in human-mouse pairs (which is more pronounced for large numbers of processors), suggesting that ParaAT is able to achieve high parallel efficiency for computationally demanding tasks.
There are four steps as follows.
Please send bugs or advice to Zhang Zhang at zhangzhang.cn at gmail dot com or zhangzhang at big dot ac dot cn.